阴道的先天性解剖结构异常。女性生殖器较为复杂的疾病之一。包括MRKH(Mayer-Rokitansky-Küster-Hauser,MRKH)综合征,阴道闭锁、阴道横隔、阴道纵隔、阴道斜隔综合征等。
阴道由副中肾管和泌尿生殖窦发育而来
阴道的先天性解剖结构异常。女性生殖器较为复杂的疾病之一。包括MRKH(Mayer-Rokitansky-Küster-Hauser,MRKH)综合征,阴道闭锁、阴道横隔、阴道纵隔、阴道斜隔综合征等。
阴道由副中肾管和泌尿生殖窦发育而来
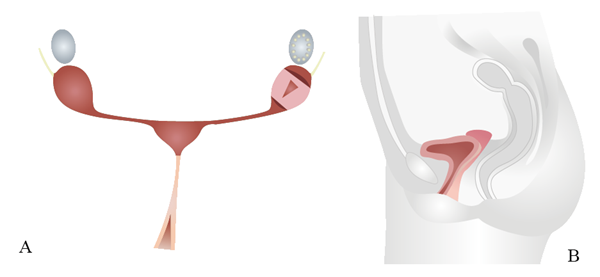
双侧副中肾管发育不全所致的一组症候。人群发病率约为1/4000~1/5000,患者表现为先天性无阴道伴无子宫或始基子宫,卵巢发育多正常。
发病机制
发病机制尚不清楚,被广为接受的假说是苗勒管-泌尿生殖窦起源。胚胎发育早期
两侧副中肾管会合后的尾端与尿生殖窦相接处未贯通或部分贯通所致的阴道发育异常。
可位于阴道内任何部位,以上、中段交界处为多见,其厚度约为1厘米。常独立存在,很少伴有泌尿系统和其他器官的异常。
临床表现和诊断
根据横隔
泌尿生殖窦未参与形成阴道下段所致的阴道发育异常。
根据阴道闭锁的解剖学特点将其分为:
①阴道下段闭锁,又称Ⅰ型阴道闭锁,阴道上段及宫颈、子宫体均正常;
②阴道完全闭锁,又称Ⅱ型阴道闭锁,多合并宫颈发育不良,子宫体发育不良或子宫畸形
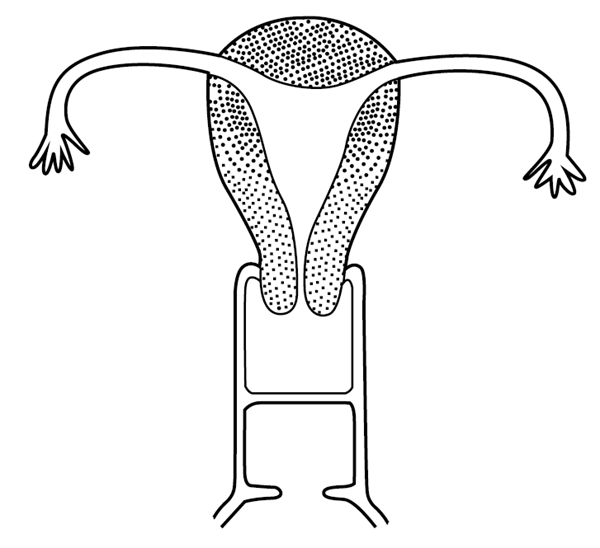
胚胎时期副中肾管下端发育异常,双侧副中肾管会和后,其中隔未消失(形成完全性阴道纵隔)或未完全消失(形成不完全性阴道纵隔)所致的女性生殖道畸形。
分为完全性阴道纵隔和不完全性阴道纵隔。前者的纵隔一般附着在阴道前、后壁的正中线上,纵向行走
表现为双子宫、双阴道及部分阴道纵隔畸形,但其阴道纵膈的末端向一侧倾斜与阴道侧壁融合成为盲端的女性生殖道畸形。
常合并斜隔一侧的肾脏缺如。系由胚胎发育的第4周早期,一侧中肾管及输尿管发育不良,无法引导该侧发育正常的苗勒氏管与对侧苗勒氏管
阴道末端未能形成与前庭相通的孔道,遗留一层薄膜成为无孔处女膜的一种处女膜解剖学变异。
分类
处女膜的解剖学变异还包括微孔处女膜、筛状处女膜、隔状处女膜(见图)。根据病因分为先天性和后天性两类。先天性处女膜闭锁是因胚胎时期副中肾管
在遗传学上有血缘关系子代的母亲。在自然状况下,则指完成子代妊娠和分娩过程的母亲。
子代的形成是一个十分复杂的过程,其可以分为相互独立的不同阶段,包括配子受精形成胚胎、子宫内妊娠过程以及出生后的抚养,而这不同过程可由同一母亲完成,也可由
生殖道与泌尿道之间的异常通道。尿液自阴道排出,不能控制。又称泌尿生殖道瘘。
按瘘管发生的部位可分为膀胱阴道瘘、尿道阴道瘘、膀胱宫颈瘘、膀胱尿道阴道瘘及输尿管阴道瘘等,以前三种为常见,占所有尿瘘的9/10。
导致尿瘘的原因很多,多
肠道与生殖道之间的异常通道,最常见的是阴道直肠瘘。
多由产伤所致,胎头在阴道内停滞过久,直肠受压坏死形成粪瘘。会阴Ⅲ度撕裂,修补后直肠未愈合或缝线穿透直肠黏膜未发现也可导致直肠阴道瘘。其次,外伤或手术直接损伤直肠,或癌瘤浸润、阴道放疗